
Introduction
On a global scale, lung cancer continues to climb in both confirmed incidence and mortality. In 2020, there were a staggering 19.3 million new cancer diagnoses and nearly 10 million cancer deaths, with lung cancer accounting for 1.79 million of those deaths, or about 18% of the global total. Lung adenocarcinoma (LUAD), as the leading subtype of lung cancer, is especially problematic due to its late diagnosis and high heterogeneity., For patients with LUAD who have missed the optimal window for radical surgery, the next line of treatment commonly includes chemotherapy, targeted therapy, immunotherapy, or combination therapies. However, resistance to chemotherapy drugs can and often does develop in these patients, thus limiting the impact of clinical interventions.,
Pemetrexed (PEM) functions by inhibiting thymidylate synthase and a spectrum of other folate-dependent enzymes, thereby interfering with the biosynthesis of nucleotides through the disruption of intracellular metabolic processes that are dependent on folates, which in turn triggers cell apoptosis., PEM is a fundamental part of the treatment for advanced LUAD, typically used in combination with other agents such as cisplatin or carboplatin, to demonstrate significant antitumor effects., However, resistance to PEM can lead to a diminished progression-free survival in patients undergoing such treatment., It is imperative to explore the molecular mechanisms behind PEM resistance in LUAD and to pinpoint therapeutic targets that can improve the clinical efficacy of PEM-based therapies. Connections between autophagy and resistance to chemotherapy in cancer have been the focus of recent research., Tong et al. reported that PEM induced a protective autophagic response in HepG2 liver cancer cells, and the sensitivity of these cells to PEM was found to be enhanced when autophagy was suppressed through the use of inhibitors or by knocking down key autophagy genes. This indicates that targeting autophagy might be a feasible strategy to improve chemosensitivity.
LncRNAs, transcripts exceeding 200 nucleotides in length, are involved in the modulation of LUAD. NEAT1, a notable lncRNA family member, is nuclear in location and is a key structural component of paraspeckles. It has been associated with the advancement of solid tumors such as gastric cancer, breast cancer, and LUAD. The precise role of lncRNA-NEAT1 in the resistance of LUAD to PEM is still to be delineated. miRNAs, non-coding single-stranded RNA molecules, are essential in the mechanisms of cancer development. They can interact with LncRNAs to influence the expression of downstream target genes. miR-379, a tumor suppressor, is a potential biomarker and therapeutic agent for non-small cell lung cancer (NSCLC). The specific function of miR-379-coding3p in cancer and its association with lncRNA-NEAT1 is not fully understood.
Incorporating bioinformatics analysis with a battery of in vitro experiments, this study has probed into the agency of lncRNA-NEAT1 in the resistance of LUAD to PEM, revealing its underlying molecular mechanisms. It has come to light that lncRNA-NEAT1 can sponge miR-379-3p and upregulate the expression of the hypoxia-inducible factor (HIF1A), thereby activating autophagy and increasing resistance to PEM. These findings enrich our understanding of the etiology of PEM resistance in LUAD and offer potential therapeutic targets for its amelioration.
Materials and methods
Bioinformatics analysis
We obtained the TCGA-LUAD dataset from TCGA (https://tcga-data.nci.nih.gov/tcga/) to perform an expression study on LncRNA-NEAT1. The interaction between LncRNA-NEAT1 and miR-379-3p was examined via StarBase (https://starbase.sysu.edu.cn/index.php). Additionally, the interaction sites of miR-379-3p with the HIF1A mRNA were explored using the TargetScan online platform (https://www.targetscan.org/vert_72/).
Cell culture
BEAS-2B (BNCC359274), a line of human bronchial epithelial cells, 293T (BNCC353535), a line of human embryonic kidney cells, and LUAD cell lines H1975 (BNCC340345), Calu-3 (BNCC359757), and A549 (BNCC337696) were acquired from BeNa Culture Collection (BNCC, China). The PEM-resistant strain A549/PEM was supplied by Meisen Chinese Tissue Culture Collections (MeisenCTCC, China). The 293 T and BEAS-2B cell lines were grown in DMEM-H (Gibco, USA) enriched with 10% FBS (Invitrogen, USA), 100 U/mL penicillin, and 100 μg/mL streptomycin (both from Invitrogen, USA). H1975 cells were nurtured in RPMI-1640 (Gibco, USA) with identical supplements. Calu-3 cells were cultured in EMEM (Gibco, USA) under the same conditions. A549 cells were maintained in F-12K (Gibco, USA), also supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. The medium for 293T cells includes 2 mM L-glutamine. The cells were incubated at 37°C in a humidified incubator with 5% CO2. Routine mycoplasma tests for all cell lines were negative.
Cell transfection
Small interfering RNA constructs si-NEAT1 and si-HIF1A, targeted at LncRNA-NEAT1 and HIF1A, along with a scrambled siRNA (si-NC), miR-379-3p mimics, and negative control mimics (mimics-NC), were all synthesized by RiboBio (China). The complete sequence of LncRNA-NEAT1 and the CDS of HIF1A were cloned into the pcDNA3.1 expression vector (Invitrogen, USA) to generate the oe-NEAT1 and oe-HIF1A plasmids, with the pcDNA3.1 empty vector serving as a control (oe-NC). A549/PEM cells were plated at 5 × 104 cells per well in six-well plates and allowed to grow overnight before the transfection process, which was carried out using Lipofectamine 3000 (Invitrogen, USA) according to the provider’s guidelines.
qRT-PCR
Total RNA from cells was isolated using TRIzol reagent (Invitrogen, USA). The RNA was reverse transcribed to cDNA for mRNA with the PrimeScript™ RT reagent Kit (Takara, Japan) and for miRNA with the Hairpin-itTM miRNAs RT-PCR Quantitation Kit (GenePharma, China). qRT-PCR was executed on the Applied Biosystems™ 7500 Real-Time PCR System, utilizing TB Green® Premix Ex Taq™ (Takara, Japan). GAPDH was used as the internal control gene for mRNA, and U6 was the control for miRNA. The 2−ΔΔCt method was applied to measure the relative expression of LncRNA-NEAT1, HIF1A, and miR-379-3p, with primer sequences provided in Table 1.

Colony formation assay
A549/PEM cells, at a density of 500 per well, were seeded into a 6-well plate for a 2-week culture period, during which the medium was changed every 3 days. Post 2 weeks, the cells were fixed using 4% paraformaldehyde (Sigma-Aldrich, USA) and stained with a 0.1% crystal violet solution (Solarbio, China). The colonies were then imaged and enumerated under a Leica light microscope (Germany).
Cell counting kit-8 (CCK-8)
CCK-8 (Beyotime, China) was employed to determine cell viability. A549/PEM cells were plated in a 96-well plate at a rate of 2 × 103 cells per well. After 0, 24, 48, and 72 h of culture, 10 μL of CCK-8 was added to each well and incubated at 37°C for 2 h. The optical density (OD) at 450 nm was then recorded using a microplate reader (BioTek, USA). The half-maximal inhibitory concentration (IC50) values were then computed employing GraphPad Prism v8.0 software (GraphPad Software Inc., USA).
PEM resistance in A549/PEM cells was appraised with the CCK-8 kit from Beyotime. The cells were plated at a density of 5 × 103 cells/well in 96-well plates and cultured for 24 h. They were treated with a spectrum of PEM concentrations (0, 25, 50, 75, 100 nM) for 48 h. A 10 μL CCK-8 solution was added to each well and incubated at 37°C for 2 h. OD at 450 nm was measured with a BioTek microplate reader. The IC50 values were calculated with GraphPad Prism v8.0 software.
Dual-luciferase reporter assay
The full-length LncRNA-NEAT1 sequence that includes the miR-379-3p binding region and its mutant form for the dual-luciferase assay were synthesized and inserted into the pmirGLO vector (Fenghbio, China), termed pmirGLO-NEAT1-wild type (WT) and pmirGLO-NEAT1-mutant (MUT).
The 3′UTR sequence of HIF1A with the miR-379-3p binding motif was amplified via PCR and integrated into the pmirGLO vector to create the pmirGLO-HIF1A-WT and pmirGLO-HIF1A-MUT vectors. We seeded 293T cells into 6-well plates and co-transfected them with the aforementioned constructs and miR-379-3p mimics or mimics-NC using Lipofectamine 3000. After a 48-h incubation, the cells were harvested, and luciferase activities were assessed with the Dual-Luciferase® Reporter Assay System (Promega, USA), normalizing firefly luciferase activity to Renilla luciferase.
RNA immunoprecipitation (RIP)
The Magna RIP® Kit by Millipore (USA) facilitated the RIP analysis in this study, with steps adhered to the manufacturer’s directions. Specifically, whole-cell lysates were derived from A549/PEM cells, to which a mixture of protease and ribonuclease inhibitors was added for a 5-min incubation. Afterward, the lysates were incubated with magnetic beads coupled with either anti-Ago2 or anti-IgG antibodies (Millipore, USA) at room temperature for 30 min. The RNA purified was quantified using qRT-PCR.
Western blot (WB)
Radioimmunoprecipitation assay (RIPA) lysis buffer with protease inhibitors (Sigma-Aldrich, USA) was used for cell lysis. The supernatant was obtained by centrifugation at 4°C, 12,000 r/min for 20 min. Protein concentrations were quantified using the bicinchoninic acid (BCA) protein assay. Proteins were resolved on SDS-PAGE gels and transferred to a PVDF membrane (Millipore, USA). After a 2-h blocking step with 5% skim milk at room temperature, the membrane was incubated with primary antibodies—anti-LC3A/B antibody (4108, CST, USA), anti-Beclin1 antibody (ab207612, Abcam, UK), and anti-GAPDH antibody (ab9485, Abcam, UK)—overnight at 4°C. The membrane was then washed with Tris-Buffered Saline Tween-20 (TBST) and incubated with the secondary antibody, goat anti-rabbit IgG H&L (HRP) (ab205718, Abcam, UK), for 2 h at room temperature. Band detection was performed with an ECL kit (OMIGET, China) using a ChemiScope 6000 imaging system (Clinx, China), and band intensity was measured using Image J software v1.8.0.
Flow cytometry
We employed flow cytometry for analyzing cell apoptosis. A549/PEM cells were digested with 0.25% Trypsin-EDTA (Beyotime, China) and rinsed with PBS (Gibco, USA) twice. The cells were stained with Annexin V-FITC and propidium iodide (PI) as per the instructions provided with the apoptosis detection kit from MULTI SCIENCES (China). After centrifugation, the cells were suspended in 1X Binding Buffer at a density of 1 × 106 cells/mL, mixed with 5 μL Annexin V-FITC and 10 μL PI, incubated at room temperature in the dark for 5 min, and analyzed using the NovoCyte flow cytometry system (Agilent, USA).
Statistical analysis
Results from three replicate experimental runs were quantitatively analyzed using GraphPad Prism v8.0 and are represented as “mean ± SD”. For the analysis of differences between two groups, t test or the Wilcoxon test was used. Differences among several groups were assessed using one-way ANOVA. The threshold for statistical significance was p < 0.05, with a 95% confidence interval.
Results
LncRNA-NEAT1 is upregulated in LUAD tissues and cells
To clarify the role and molecular mechanisms of LncRNA-NEAT1 in LUAD, we initially analyzed its expression levels. The TCGA dataset analysis indicated that LncRNA-NEAT1 is markedly overexpressed in lung cancer tissues compared to normal lung tissues (Figure 1(a)). Moreover, qRT-PCR analysis of various cell lines showed that compared to human bronchial epithelial cell line (BEAS-2B), LncRNA-NEAT1 was expressed at higher levels in LUAD cell lines (A549, H1975, and Calu-3) (Figure 1(b)).
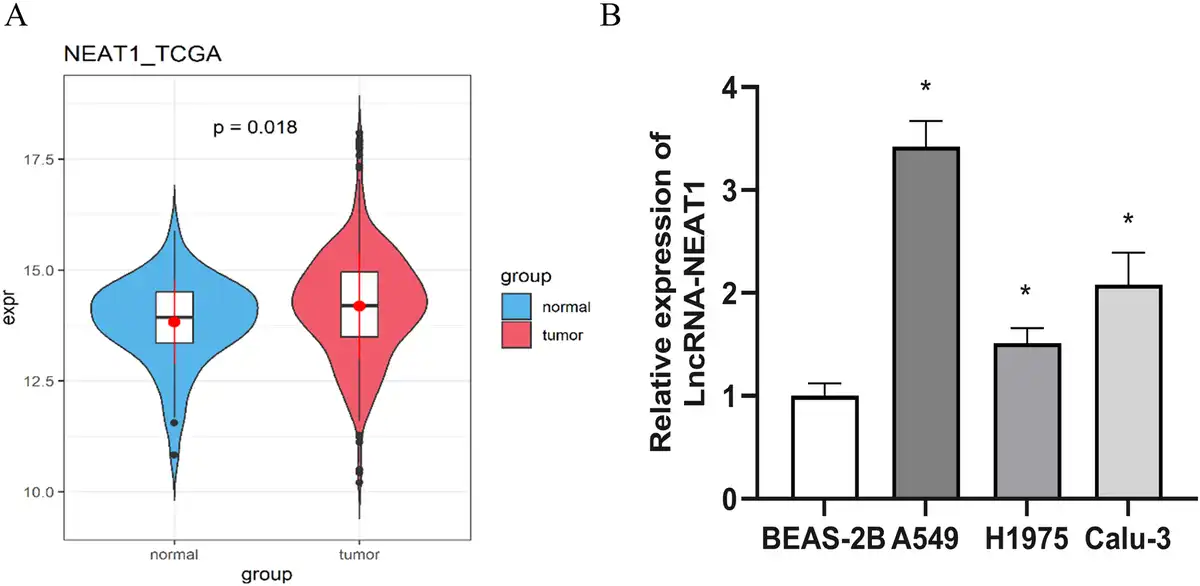
Figure 1
High expression of LncRNA-NEAT1 in LUAD. (a): Expression levels of LncRNA-NEAT1 in normal and tumor tissues from LUAD patients in the TCGA database; (b): qRT-PCR detection of LncRNA-NEAT1 expression levels in bronchial epithelial cells (BEAS-2B) and LUAD cell lines (A549, H1975, and Calu-3). * denotes p < 0.05.
LncRNA-NEAT1 promotes the proliferation of LUAD cells
To determine if there is a link between LncRNA-NEAT1 and resistance to PEM in LUAD, we began by using qRT-PCR to evaluate LncRNA-NEAT1 expression in PEM-resistant cell lines and their parental cells. A549, which has a high intrinsic expression level of LncRNA-NEAT1, was chosen for subsequent analysis. A substantial increase in LncRNA-NEAT1 expression was identified in A549/PEM cells compared to A549 cells (Figure 2(a)), hinting that the dysregulation of LncRNA-NEAT1 could be associated with PEM resistance. Cellular function experiments were then conducted to assess the contribution of LncRNA-NEAT1 to the acquisition of PEM resistance, qRT-PCR analysis confirmed the knockdown efficiency of LncRNA-NEAT1, as LncRNA-NEAT1 expression was significantly downregulated in si-NEAT1-transfected A549/PEM cells (Figure 2(b)). CCK-8-based cell proliferation tests and cloning assays indicated that the depletion of LncRNA-NEAT1 notably suppressed the growth of A549/PEM cells (Figure 2(c) and (d)). Flow cytometry analysis revealed that the introduction of si-NEAT1 into A549/PEM cells resulted in a marked enhancement of PEM-induced apoptosis (Figure 2(e)). We measured the absorbance using CCK-8 after treating the cells with different concentrations (0, 25, 50, 75, 100 nM) of PEM, finding that the IC50 values were impressively reduced with LncRNA-NEAT1 knockdown, suggesting heightened sensitivity to PEM (Figure 2(f)). These data collectively imply that LncRNA-NEAT1 aids in the development of PEM resistance in LUAD cells.
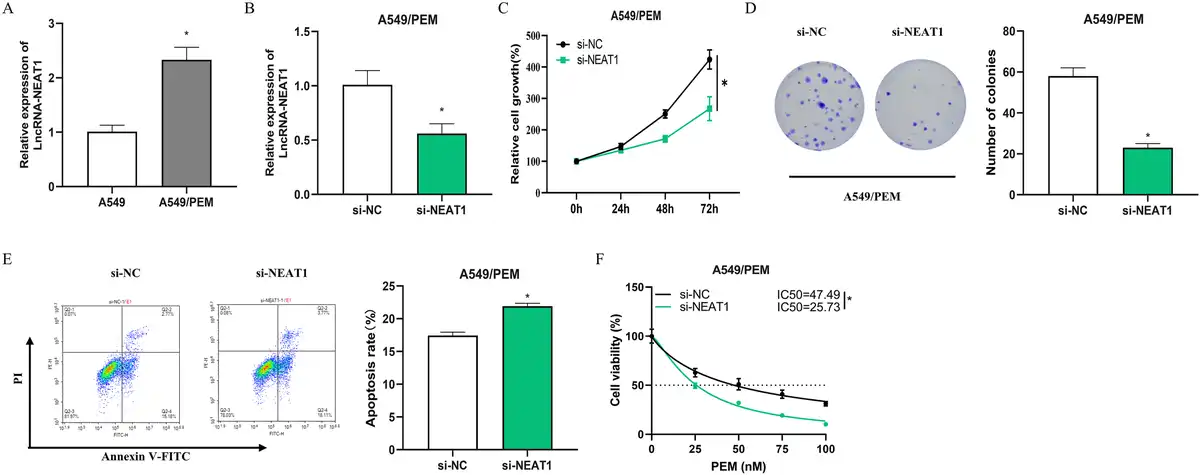
Figure 2
Suppression of lncRNA-NEAT1 diminishes A549/PEM cell resistance to PEM (a): qRT-PCR analyzed LncRNA-NEAT1 expression in A549 cells and PEM-resistant A549 cells (A549/PEM); (b): qRT-PCR assessed the transfection rates for si-NC or si-NEAT1; (c-d): CCK-8 proliferation and cloning assays measured cell growth across groups; (e): Flow cytometric analysis of apoptosis in different groups; (f): CCK-8 assay was conducted to evaluate the inhibitory effects of PEM on A549/PEM cell growth and to determine the IC50. * denotes p < 0.05.
LncRNA-NEAT1 sponges miR-379-3p and dampens its expression in LUAD
To extend our study on the mechanisms of LncRNA-NEAT1 in LUAD, we used bioinformatics tools to forecast the potential targets of LncRNA-NEAT1. A binding site for LncRNA-NEAT1 was identified within the miR-379-3p sequence (Figure 3(a)). After verifying the transfection efficiency of mimics-NC and miR-379-3p mimics (Figure 3(b)), we performed dual-luciferase and RIP to scrutinize the interaction between LncRNA-NEAT1 and miR-379-3p (Figure 3(c) and (d)). The dual-luciferase results indicated a notable decrease in luciferase activity in 293T cells with the NEAT1-WT construct, in contrast to those with NEAT1-MUT, upon miR-379-3p mimics transfection (Figure 3(c)). Furthermore, RIP data indicated that the anti-Ago2 antibody could substantially enrich LncRNA-NEAT1 and miR-379-3p, as opposed to the IgG control (Figure 3(d)). To further appraise the regulatory influence of LncRNA-NEAT1 on miR-379-3p, qRT-PCR was applied to evaluate miR-379-3p levels in A549/PEM cells transfected with si-NC or si-NEAT1. A marked elevation in miR-379-3p expression was identified in cells treated with si-NEAT1 (Figure 3(e)). The findings altogether suggest that LncRNA-NEAT1 binds to and curbs the expression of miR-379-3p.
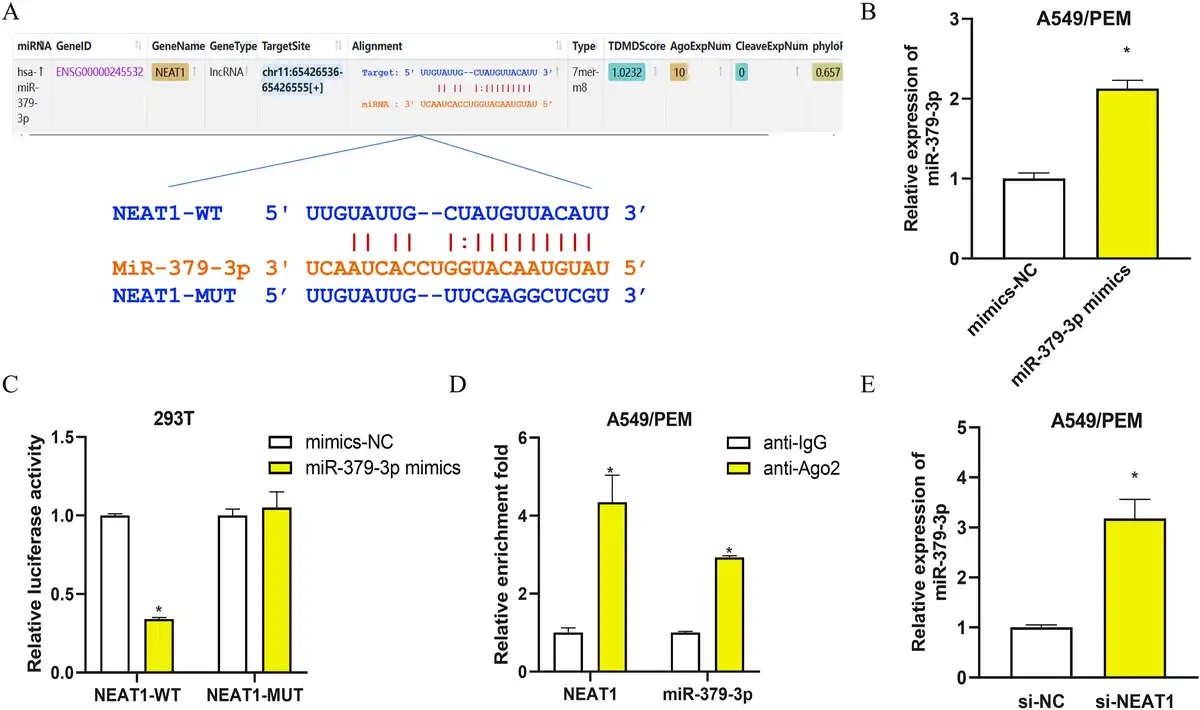
Figure 3
LncRNA-NEAT1 binds to and represses the expression of miR-379-3p in LUAD cells (a): Schematic representation of the binding site between LncRNA-NEAT1 and miR-379-3p, along with the mutation design based on the binding site; (b): qRT-PCR detection of transfection efficiency for mimics-NC or miR-379-3p mimics; (c): Dual-luciferase assay for analyzing the interaction between LncRNA-NEAT1 and miR-379-3p; (d): RIP analysis performed with anti-Ago2 antibody in A549/PEM cell extracts and assessment of immunoprecipitated RNA levels by qRT-PCR; (e): qRT-PCR detection of miR-379-3p expression in A549/PEM cells transfected with si-NC or si-NEAT1. * denotes p < 0.05.
Impact of miR-379-3p on the resistance of LUAD cells to PEM by inhibiting HIF1A and downregulating autophagy
MiRNAs typically participate in the post-transcriptional regulation of gene expression by binding to their target mRNAs. Bioinformatics analysis has suggested a potential binding affinity between miR-379-3p and the 3′UTR of the HIF1A gene (Figure 4(a)). We have experimentally verified this interaction using dual-luciferase assays and RIP (Figure 4(b) and (c)). The luciferase activity was significantly lower in cells with the wild-type HIF1A sequence when treated with miR-379-3p mimics, as compared to those with the mutated sequence (Figure 4(b)). RIP results showed that the anti-Ago2 antibody could substantially enrich miR-379-3p and HIF1A, in contrast to the IgG control (Figure 4(c)). Additionally, the mRNA levels of HIF1A were found to be significantly decreased in A549/PEM cells following the transfection of miR-379-3p mimics (Figure 4(d)). These results indicate that miR-379-3p may be downregulating HIF1A expression by interacting with its 3′UTR.
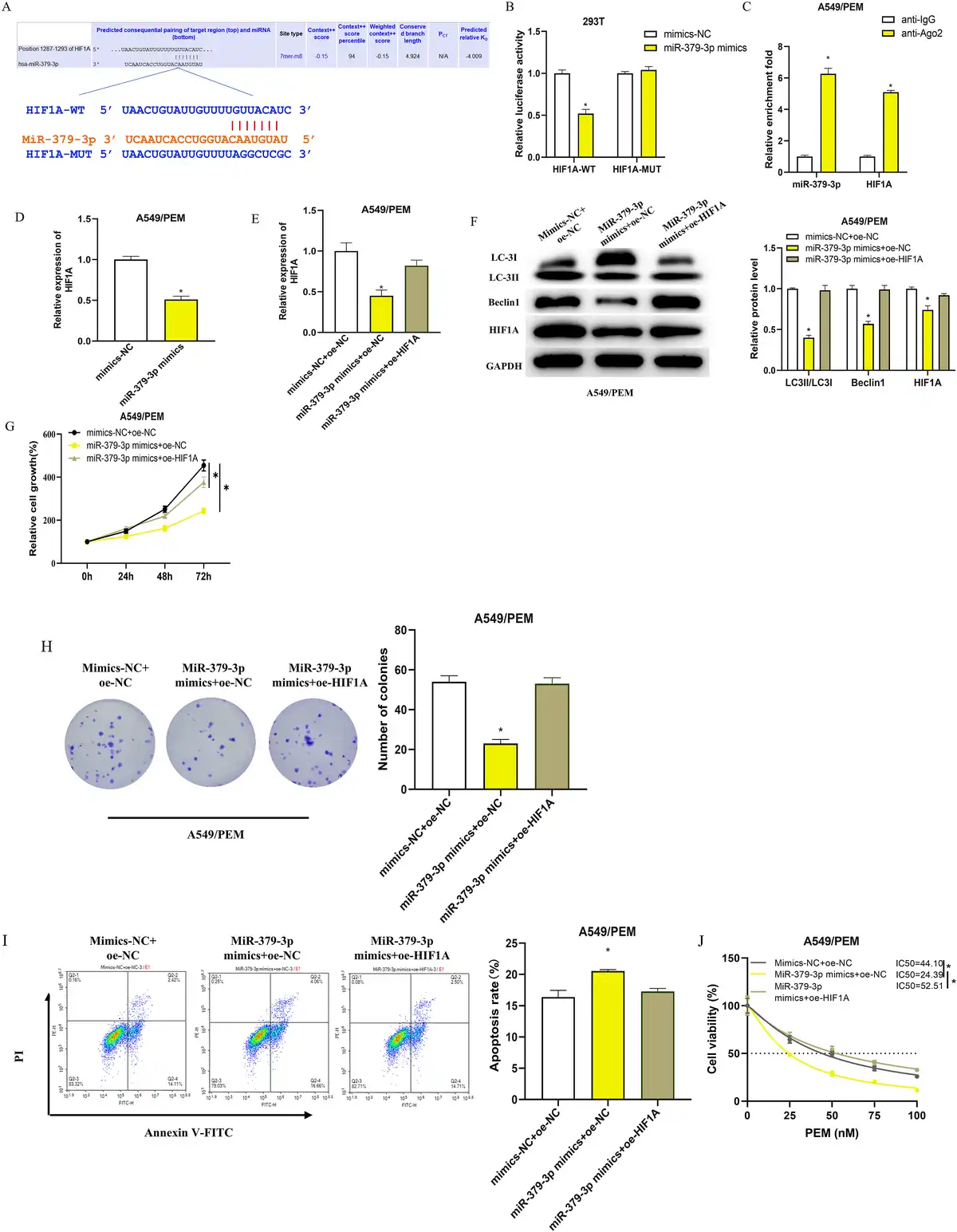
Figure 4
Impact of miR-379-3p on the resistance of LUAD cells to PEM by inhibiting HIF1A and downregulating autophagy (a): Illustration of the miR-379-3p binding site on HIF1A and the mutation design predicated on this interaction; (b): Dual-luciferase assay dissected the relationship between miR-379-3p and the 3′UTR of HIF1A; (c): RIP analysis using an anti-Ago2 antibody on A549/PEM cell extracts, followed by qRT-PCR quantification of the RNA precipitated; (d): qRT-PCR analysis of HIF1A mRNA levels in A549/PEM cells following transfection with either mimics-NC or miR-379-3p mimics; (e): qRT-PCR quantified HIF1A mRNA in cells from the mimics NC + oe-NC, miR-379-3p mimics + oe-NC, or miR-379-3p mimics + oe-HIF1A groups; F: WB analysis of autophagy markers (LC3II/LC3I, Beclin1) and HIF1A protein levels across different cell groups; (g-h): CCK-8 and colony formation assays assessed cell growth in various groups; (i): Apoptosis assessment in different groups using flow cytometry; (j): CCK-8 assay measured the impact of PEM on A549/PEM cell viability, with the IC50 values calculated. * denotes p < 0.05.
Evidence suggests that HIF1A can foster cancer cell resistance to drugs by promoting autophagy, as documented in research articles.– We theorized that miR-379-3p could diminish the autophagy-induced drug resistance in LUAD cells by inhibiting HIF1A expression. To validate this, we created distinct cell groups including mimics NC + oe-NC, miR-379-3p mimics + oe-NC, and miR-379-3p mimics + oe-HIF1A, and subsequently measured transfection efficacy via qRT-PCR. The miR-379-3p mimics + oe-NC group exhibited a marked reduction in HIF1A levels compared to the mimics NC + oe-NC group, whereas the miR-379-3p mimics + oe-HIF1A group saw a return to baseline HIF1A expression (Figure 4(e)). Additionally, the diminished HIF1A protein levels led to a remarkable reduction in the autophagy markers LC3II/LC3I ratio and Beclin one protein expression in A549/PEM cells (Figure 4(f)). Through CCK-8 and colony formation assays, we discerned that cell proliferation in the miR-379-3p mimics + oe-NC group was substantially impaired relative to the mimics NC + oe-NC group while the proliferative capacity of cells in the miR-379-3p mimics + oe-HIF1A group was comparable to that in the control group (Figure 4(g) and (h)). Conversely, apoptosis was most evident in the miR-379-3p mimics + oe-NC group (Figure 4(i)). By measuring the absorbance of each group with the CCK-8 method and calculating the IC50 values, we found that transfection with miR-379-3p mimics + oe-NC notably reduced the IC50 of PEM for A549/PEM cells (Figure 4(j)). Collectively, our findings suggest that miR-379-3p attenuates the autophagy levels and consequently the drug resistance of LUAD cells to PEM by inhibiting HIF1A expression.
LncRNA-NEAT1 drives autophagy and promotes PEM resistance in LUAD cells via the miR-379-3p/HIF1A axis
To explore the potential role of LncRNA-NEAT1 in modulating the miR-379-3p/HIF1A axis and thereby influencing PEM resistance in LUAD, we developed four A549/PEM cell groups: oe-NC, oe-NEAT1, oe-NEAT1 + miR-379-3p mimics, and oe-NEAT1 + si-HIF1A. By employing qRT-PCR to quantify the expression of LncRNA-NEAT1, miR-379-3p, and HIF1A, we discovered that elevated LncRNA-NEAT1 levels led to a significant downregulation of miR-379-3p, a decrease that was neutralized by miR-379-3p mimics. In addition, the overexpression of LncRNA-NEAT1 substantially boosted HIF1A expression, an effect that was negated by miR-379-3p mimics or si-HIF1A (Figure 5(a)). We then proceeded with WB experiments to evaluate the expression of autophagy-associated proteins (LC3II/LC3I, Beclin1) and HIF1A in the different cell groups (Figure 5(b)). The findings revealed that the levels of these proteins in the oe-NEAT1 + miR-379-3p mimics and oe-NEAT1 + si-HIF1A groups were more aligned with the oe-NC group. On the other hand, the oe-NEAT1 group displayed a conspicuous increase in HIF1A protein levels, as well as a notable increase in the LC3II/LC3I ratio and Beclin1 protein levels, implying that an increase in HIF1A led to the onset of autophagy. The oe-NEAT1-induced enhancement of cell proliferation was significantly reversed by miR-379-3p mimics or si-HIF1A (Figure 5(c) and (d)). Furthermore, the apoptosis-inhibiting effect of oe-NEAT1 was also reduced by miR-379-3p mimics or si-HIF1A (Figure 5(e)). Utilizing the CCK-8 assay to determine the absorbance of the groups and to compute the IC50 values, we found that the overexpression of NEAT1 notably elevated the IC50 of PEM for A549/PEM cells. In contrast, the introduction of miR-379-3p mimics or si-HIF1A caused a decline in the IC50, thus diminishing the resistance of A549/PEM cells to PEM (Figure 5(f)). The aforementioned findings suggest that the LncRNA-NEAT1/miR-379-3p/HIF1A pathway may confer resistance to PEM in LUAD cells through the induction of autophagy.
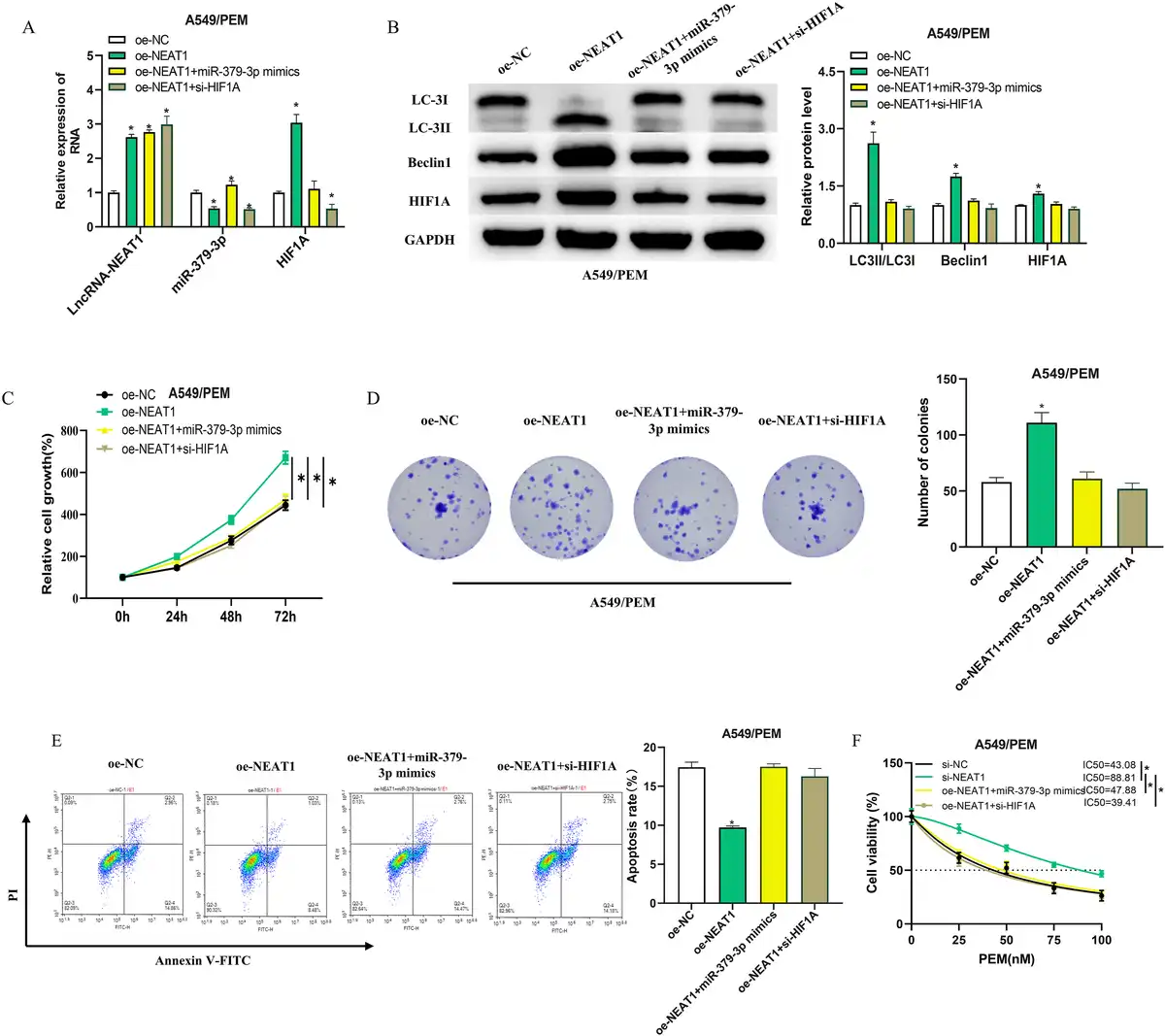
Figure 5
LncRNA-NEAT1 drives autophagy and promotes PEM resistance in LUAD cells via the miR-379-3p/HIF1A axis. (a): qRT-PCR assessment of LncRNA-NEAT1, miR-379-3p, and HIF1A expression in four categories of A549/PEM cells: oe-NC, oe-NEAT1, oe-NEAT1 + miR-379-3p mimics, and oe-NEAT1 + si-HIF1A; (b): WB quantification of autophagy-related proteins (LC3II/LC3I, Beclin1) and HIF1A in different cell groupings; (c-d): CCK-8 gauged the proliferative capacity in different groups; (e): Apoptosis rate measurement in different cell groupings using flow cytometry; (f): CCK-8 measured the impact of PEM on A549/PEM cell viability, with the IC50 values calculated. * denotes p < 0.05.
Discussion
Chemotherapy centered around PEM typically yields positive outcomes in the majority of LUAD patients; however, the development of drug resistance is increasingly becoming a formidable challenge in oncology practice. In our research, we probed into the role of lncRNA-NEAT1 in the development of resistance to PEM in LUAD and the underlying control systems. With bioinformatics data and in vitro experimental evidence, we discovered that lncRNA-NEAT1 is more prevalent in LUAD tissues or cells than in the normal tissues adjacent to the carcinoma or in the cell lines of the pulmonary bronchial epithelium. Our data also pointed to a higher expression of lncRNA-NEAT1 in PEM-resistant strains of A549 cells compared to their original cell line. The IC50 values indicated that lncRNA-NEAT1 boosted the resistance of A549 cells to PEM, a process that was brought about by engaging with the miR-379-3p/HIF1A axis and stimulating autophagy.
Attention to lncRNAs has grown in recent years because of their key roles in the carcinogenic process. Yet, the biological significance of the majority of lncRNAs is still not fully elucidated, particularly in terms of their connection to resistance to cancer therapies. In our research, we developed two A549/PEM cell models, si-NC and si-NEAT1, and through a comprehensive set of cellular experiments, we determined that the suppression of lncRNA-NEAT1 notably curtailed the growth capacity of A549/PEM, boosted the incidence of apoptosis, and lessened the maximum concentration of PEM necessary for half-maximal inhibition in A549/PEM cells. These results imply that LncRNA-NEAT1 serves as a promoter of oncogenesis in LUAD and enhances the resistance to PEM. Parallel findings were reported by Mu et al.,– who demonstrated that the suppression of LncRNA-NEAT1 alleviated the sorafenib resistance in NSCLC, presenting with reduced cell proliferation and increased apoptotic rates. Our study contributes further evidence to the role of LncRNA-NEAT1 in tumor resistance and indicates the possibility of LncRNA-NEAT1 as a target for modulating PEM resistance in LUAD.
The axis of lncRNA-miRNA-mRNA interaction is acknowledged as a vital component in the pathology of diverse cancers. LncRNAs, within this framework, can function as competitive endogenous RNAs (ceRNAs), sequestering miRNAs and thus affecting the regulation of target gene expression downstream., Zhu et al. found that LncRNA-NEAT1 sponges miR-23a-3p to elevate FOXA1 levels, enhancing the resistance of breast cancer to paclitaxel. Shen et al. demonstrated the contribution of the LINC00518/miR-335-3p/CTHRC1 axis to the promotion of proliferation and metastasis in LUAD.. Consequently, we performed bioinformatics analysis to seek out possible targets of lncRNA-NEAT1. We identified potential binding sites for miR-379-3p within lncRNA-NEAT1 and validated their interaction via dual-luciferase reporter assay and RIP. qRT-PCR data revealed that silencing lncRNA-NEAT1 in A549/PEM cells notably increased the levels of miR-379-3p, indicating that lncRNA-NEAT1 may sponge miR-379-3p and reduce its expression in LUAD cells. Prior research has shown that miR-379 exhibits tumor-suppressive activity across multiple tissues such as the brain, breast, lung, and liver. The miR-379 family includes two mature variants, miR-379-5p and miR-379-3p. miR-379-5p is known to suppress cisplatin resistance, curb cell growth, and encourage cell death in lung cancer.– In stark contrast, miR-379-3p has received minimal research attention. Thus, our research contributes to filling this gap in the literature.
We proceeded to forecast the downstream targets of miR-379-3p and discovered complementary binding sites between miR-379-3p and the 3′UTR of HIF1A. The interaction was validated through dual-luciferase reporter assay, RIP, and qRT-PCR, demonstrating the capacity of miR-379-3p to repress HIF1A expression. Rescue assays revealed that the introduction of miR-379-3p mimics reduced the drug resistance of A549/PEM to PEM by decreasing HIF1A levels, an effect that was counteracted by the overexpression of NEAT1. HIF1A has been noted for its involvement in chemoresistance among various tumor cells by triggering autophagy, as evidenced in earlier studies., Indeed, a plethora of research points to the connection between autophagy and chemoresistance in cancer. Li et al. illuminated how OTUD6B-AS1/miR-26a-5p/MTDH contributes to the emergence of paclitaxel resistance in triple-negative breast cancer through heightened autophagy and the inhibition of DNA damage response-driven genomic instability. Hwang et al. indicate that curbing autophagy induced by PEM and simvastatin can intensify apoptosis in NSCLC cells. Hence, the suppression of autophagy may represent a significant pathway to bolster the chemotherapeutic responsiveness of cancer cells. Within our research, it was found that the downregulation of HIF1A in A549/PEM cells by miR-379-3p mimics led to a decrease in the LC3II/LC3I ratio and Beclin1 protein levels, signaling a reduction in autophagic activity. Simultaneously, the resistance of A549/PEM cells to PEM was diminished. Yet, when miRNA-379-3p expression was suppressed by lncRNA-NEAT1, these effects were counteracted. Our findings point out that the lncRNA-NEAT1/miR-379-3p/HIF1A pathway may increase the resistance of LUAD cells to PEM by facilitating autophagy. However, the specific mechanism in which autophagy enhances the resistance of these cells to PEM is not well understood and deserves further exploration.
To synthesize our work, the lncRNA-NEAT1/miR-379-3p/HIF1A axis has been identified as a pivotal regulatory signal in the resistance of LUAD to PEM. We have demonstrated that lncRNA-NEAT1 sponges miR-379-3p, thus sustaining the expression of HIF1A, and that the overexpression of HIF1A, by promoting autophagy, augment the resistance of LUAD cells to PEM. Therefore, targeting the lncRNA-NEAT1/miR-379-3p/HIF1A axis or inhibiting autophagy could be effective treatments for patients with PEM-resistant LUAD, a proposition that needs validation through clinical and animal studies.
Author contribution All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agreed to be accountable for all aspects of the work.
Declaration of conflicting interests The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
- 1. Bade BC, Dela Cruz CS. Lung cancer 2020: epidemiology, etiology, and prevention. Clin Chest Med 2020; 41: 1–24.
- 2. Ferlay J, Colombet M, Soerjomataram I, et al. Cancer statistics for the year 2020: an overview. Int J Cancer 2021; 149.
- 3. Barta JA, Powell CA, Wisnivesky JP. Global epidemiology of lung cancer. Ann Glob Health 2019; 85.
- 4. Seguin L, Durandy M, Feral CC. Lung adenocarcinoma tumor origin: a guide for personalized. Medicine. Cancers (Basel) 2022; 14.
- 5. Zhang H, Wang SQ, Wang L, et al. m6A methyltransferase METTL3-induced lncRNA SNHG17 promotes lung adenocarcinoma gefitinib resistance by epigenetically repressing LATS2 expression. Cell Death Dis 2022; 13: 657.
- 6. Zhang K, Chen J, Li C, et al. Exosome-mediated transfer of SNHG7 enhances docetaxel resistance in lung adenocarcinoma. Cancer Lett 2022; 526: 142–154.
- 7. Bodor JN, Kasireddy V, Borghaei H. First-line therapies for metastatic lung adenocarcinoma without a driver mutation. J Oncol Pract 2018; 14: 529–535.
- 8. Vora PA, Patel R, Dharamsi A. Pemetrexed - first-line therapy for non-squamous non-small cell lung cancer: a review of patent literature. Recent Pat Anti-Cancer Drug Discov 2021; 16: 333–349.
- 9. Dong X, Huang Y, Yi T, et al. Intrapleural infusion of tumor cell-derived microparticles packaging methotrexate or saline combined with pemetrexed-cisplatin chemotherapy for the treatment of malignant pleural effusion in advanced non-squamous non-small cell lung cancer: a double-blind, randomized, placebo-controlled study. Front Immunol 2022; 13: 1002938.
- 10. Noronha V, Patil VM, Joshi A, et al. Gefitinib versus gefitinib plus pemetrexed and carboplatin chemotherapy in EGFR-mutated lung cancer. J Clin Oncol 2020; 38: 124–136.
- 11. Park S, Kim JY, Lee SH, et al. KRAS G12C mutation as a poor prognostic marker of pemetrexed treatment in non-small cell lung cancer. Korean J Intern Med (Engl Ed) 2017; 32: 514–522.
- 12. Chen Y, Sun Y, Zhao W, et al. Elevated SRC3 expression predicts pemetrexed resistance in lung adenocarcinoma. Biomed Pharmacother 2020; 125: 109958.
- 13. Huang T, Xu Z, Cao L, et al. Autophagy regulation by microRNAs in chemotherapy resistance (Review). Oncol Rep 2020; 44: 791–794.
- 14. Zamame Ramirez JA, Romagnoli GG, Kaneno R. Inhibiting autophagy to prevent drug resistance and improve anti-tumor therapy. Life Sci 2021; 265: 118745.
- 15. Tong Y, Huang H, Pan H. Inhibition of MEK/ERK activation attenuates autophagy and potentiates pemetrexed-induced activity against HepG2 hepatocellular carcinoma cells. Biochem Biophys Res Commun 2015; 456: 86–91.
- 16. Xie Y, Zheng ZW, He HT, et al. LncRNA NEAT1 induces autophagy through the miR-128-3p/ADAM28 axis to suppress apoptosis of nonsmall-cell lung cancer. Kaohsiung J Med Sci 2022; 38: 933–949.
- 17. Yamazaki T, Souquere S, Chujo T, et al. Functional domains of NEAT1 architectural lncRNA induce paraspeckle assembly through phase separation. Mol Cell 2018; 70: 1038–1053.
- 18. Xu Y, Li Y, Qiu Y, et al. LncRNA NEAT1 promotes gastric cancer progression through miR-17-5p/tgfβr2 Axis up-regulated angiogenesis. Front Cell Dev Biol 2021; 9: 705697.
- 19. Liu X, Yao W, Xiong H, et al. LncRNA NEAT1 accelerates breast cancer progression through regulating miR-410-3p/ CCND1 axis. Cancer Biomarkers 2020; 29: 277–290.
- 20. Xiong DD, Li ZY, Liang L, et al. The LncRNA NEAT1 accelerates lung adenocarcinoma deterioration and binds to mir-193a-3p as a competitive endogenous RNA. Cell Physiol Biochem 2018; 48: 905–918.
- 21. Hussen BM, Hidayat HJ, Salihi A, et al. MicroRNA: a signature for cancer progression. Biomed Pharmacother 2021; 138: 111528.
- 22. Zhou RS, Zhang EX, Sun QF, et al. Integrated analysis of lncRNA-miRNA-mRNA ceRNA network in squamous cell carcinoma of tongue. BMC Cancer 2019; 19: 779.
- 23. Liu B, Wang Z, Cheng S, et al. miR-379 inhibits cell proliferation and epithelial-mesenchymal transition by targeting CHUK through the NF-κB pathway in non-small cell lung cancer. Mol Med Rep 2019; 20: 1418–1428.
- 24. Ali SZ, Langden SSS, Munkhzul C, et al. Regulatory mechanism of MicroRNA expression in cancer. Int J Mol Sci 2020; 21.
- 25. Mao X, Nanzhang, Xiao J, et al. Hypoxia-induced autophagy enhances cisplatin resistance in human bladder cancer cells by targeting hypoxia-inducible factor-1α. J Immunol Res 2021; 2021: 8887437.
- 26. Zhang X, Qi Z, Yin H, et al. Interaction between p53 and Ras signaling controls cisplatin resistance via HDAC4- and HIF-1α-mediated regulation of apoptosis and autophagy. Theranostics 2019; 9: 1096–1114.
- 27. Liu YF, Luo D, Li X, et al. PVT1 knockdown inhibits autophagy and improves gemcitabine sensitivity by regulating the MiR-143/HIF-1α/VMP1 Axis in pancreatic cancer. Pancreas 2021; 50: 227–234.
- 28. Tan YT, Lin JF, Li T, et al. LncRNA-mediated posttranslational modifications and reprogramming of energy metabolism in cancer. Cancer Commun 2021; 41: 109–120.
- 29. Rajagopal T, Talluri S, Akshaya RL, et al. HOTAIR LncRNA: a novel oncogenic propellant in human cancer. Clin Chim Acta 2020; 503: 1–18.
- 30. Mu L, Zhao H, Yang Y, et al. Long noncoding RNA NEAT1 aggravates sorafenib-resistance in non-small cell lung cancer via regulating miRNA-335/c-Met. J buon 2021; 26: 345–352.
- 31. Wang JY, Yang Y, Ma Y, et al. Potential regulatory role of lncRNA-miRNA-mRNA axis in osteosarcoma. Biomed Pharmacother 2020; 121: 109627.
- 32. Zeng X, Xiao J, Bai X, et al. Research progress on the circRNA/lncRNA-miRNA-mRNA axis in gastric cancer. Pathol Res Pract 2022; 238: 154030.
- 33. Zhu L, Wang F, Fan W, et al. lncRNA NEAT1 promotes the Taxol resistance of breast cancer via sponging the miR-23a-3p-FOXA1 axis. Acta Biochim Biophys Sin 2021; 53: 1198–1206.
- 34. Shen R, Cai X, Shen D, et al. Long noncoding RNA LINC00518 contributes to proliferation and metastasis in lung adenocarcinoma via the miR-335-3p/CTHRC1 Axis. Cell Death Dis 2022; 8: 98.
- 35. Ghafouri-Fard S, Shaterabadi D, Abak A, et al. An update on the role of miR-379 in human disorders. Biomed Pharmacother 2021; 139: 111553.
- 36. Wang L, Wang D, Xu Z, et al. Circ_0010235 confers cisplatin resistance in lung cancer by upregulating E2F7 through absorbing miR-379-5p. Thorac Cancer 2023; 14: 1946–1957.
- 37. Jiang Y, Zhu P, Gao Y, et al. miR-379-5p inhibits cell proliferation and promotes cell apoptosis in non-small cell lung cancer by targeting β-arrestin-1. Mol Med Rep 2020; 22: 4499–4508.
- 38. Li PP, Li RG, Huang YQ, et al. LncRNA OTUD6B-AS1 promotes paclitaxel resistance in triple negative breast cancer by regulation of miR-26a-5p/MTDH pathway-mediated autophagy and genomic instability. Aging (Albany NY) 2021; 13: 24171.
- 39. Hwang KE, Kim YS, Jung JW, et al. Inhibition of autophagy potentiates pemetrexed and simvastatin-induced apoptotic cell death in malignant mesothelioma and non-small cell lung cancer cells. Oncotarget 2015; 6: 29482.